 Shaoying Li1†
Shaoying Li1† Jianchun He1†
Jianchun He1† Liming Chu2†
Liming Chu2† Shuai Ren2
Shuai Ren2 Wenzhi He1Xiaoyan Ma1
Wenzhi He1Xiaoyan Ma1 Yanchao Wang1Mincong Zhang1Lingyin Kong2Bo Liang3*
Yanchao Wang1Mincong Zhang1Lingyin Kong2Bo Liang3* Qing Li1*1Department of Obstetrics and Gynecology, Experimental Department of Institute of Obstetrics and Gynecology, The Third Affiliated Hospital of Guangzhou Medical University, Key Laboratory for Major Obstetric Diseases of Guangdong Province, Guangzhou, China2Basecare Medical Device Co., Ltd, Suzhou, China3State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic and Developmental Sciences, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
Qing Li1*1Department of Obstetrics and Gynecology, Experimental Department of Institute of Obstetrics and Gynecology, The Third Affiliated Hospital of Guangzhou Medical University, Key Laboratory for Major Obstetric Diseases of Guangdong Province, Guangzhou, China2Basecare Medical Device Co., Ltd, Suzhou, China3State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic and Developmental Sciences, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, ChinaHemophilia A (HA, OMIM#306700) is an X-linked recessive bleeding disorder caused by the defects in the F8 gene, which encodes coagulation factor VIII (FVIII). Intron 22 inversion (Inv22) is found in about 45% of patients with severe hemophilia A. Here, we reported a male without obvious hemophilia A phenotype but bearing an inherited segmental variant duplication encompassing F8 as well as Inv22. The duplication was approximately 0.16 Mb and involved from exon 1 to intron 22 of F8. This partial duplication and Inv22 in F8 was first found in the abortion tissue of his older sister with recurrent miscarriage. The genetic testing of his family revealed that his phenotypically normal older sister and mother also had this heterozygous Inv22 and a 0.16 Mb partial duplication of F8, while his father was genotypically normal. The integrity of the F8 gene transcript was verified by sequencing of the adjacent exons at the inversion breakpoint, which explained why this male had no phenotype for hemophilia A. Interestingly, although he had no significant hemophilia A phenotype, the expression of C1QA in his mother, sister, and the male subject was only about half of that in his father and normal population. Our report broadens the mutation spectrum of F8 inversion and duplication and its pathogenicity in hemophilia A.
IntroductionHemophilia A (coagulation factor VIII(FVIII) deficiency), which is caused by the mutation in F8 gene and leads to abnormal production or function of FVIII protein, is the most common clinical hereditary hemorrhagic disease, with an incidence of about 1/5,000 in males (Lenting et al., 1998). F8 gene locates at the end of the long arm of X chromosome (Xq28), including 25 introns and 26 exons, with a total length of about 186 kb (Gitschier et al., 1984; Freije and Schlessinger, 1992). Intron 22 contains a 9.5-kb sequence Int22h-1, which is about 5.78 kb downstream of exon 22, and there are two homologous repeats (Int22h-2 and Int22h-3) of about 497 kb upstream of F8 gene. Intron 22 inversion (Inv22) is caused by this homologous recombination of Int22h-1 and Int22h-2/Int22h-3 (Lakich et al., 1993). A broad spectrum of mutations is known to cause hemophilia A, and Inv22 is the main pathogenic mechanism, accounting for about 45% of severe hemophilia A (Gouw et al., 2012; Lu et al., 2020). Large duplications of F8 are relatively uncommon, compared to the small genetic variants, and account for approximately 0.07% (Lannoy and Hermans, 2016). F8 duplications have varying clinical implications, from benign to severe HA phenotypes, depending on the location of insertion of the gained region (Zimmermann et al., 2010; Lannoy et al., 2013; Lannoy et al., 2015; Lannoy et al., 2019).
Here, we described a Chinese pedigree presented without hemophilia A but harboring partial duplication and Inv22 in F8. To the best of our knowledge, this is the first report of such complex structural variation of F8.
Materials and methodsCNV analysis was based on low-depth whole-genome sequencing. CNV-Seq was conducted by using the DA8600 sequencer (Daan Gene), and tMAP (version 4.6), Picard (version 2.18.17), LOWESS regression, and circular binary segmentation were used for CNV analysis (Liu et al., 2020). Inv22 assay was performed using the F8 specific inversion panel. Briefly, we performed PCR amplification by three specific primers (B: 5′- CCCCAAACTATAACCAGCACCTTGAACTTCCCCTCTCATA-3’; P: 5′- GCCCTGCCTGTCCATTACACTGATGACATTATGCTGAC-3’; and Q: 5′- GGCCCTACAACCATTCTGCCTTTCACTTTCAGTGCAATA-3′). The P&Q combination could amplify a 12-kb amplification product in samples without Inv22, while the B&Q combination could amplify an 11-kb amplification product in samples with Inv22. Detailed experimental procedures were described previously (Mahmoud Abu Arra et al., 2020).
Total RNA was isolated from each blood sample of the family members, and RNA-seq was conducted using the Illumina NovaSeq platform with 2 × 150 bp pair-end reads. FastQC (v0.11.5) was used for quality control of raw data. Adaptor sequences were removed using Cutadapt (v3.0). The proportion of ribosomal RNA (rRNA) in reads was assessed using CollectRNASeqMetrics of GATK (v4.1.2.0).
Data were aligned to the human reference genome (GRCh37) using STAR (v2.7.7a) (Dobin et al., 2013). HTseq-count (v0.12.4) was used to extract the number of each gene reads, and DEGSeq was used for differential expression analysis. Criteria for differential expression gene screening were q-value ≤0.05 and log fold change ≥1. Reference databases for male and female samples were constructed as follows: sample IDs of whole-blood samples were obtained from the file (GTEx_Analysis_v8_Annotations_SampleAttributesDS.txt); gene reads of the corresponding samples were obtained from the file (GTEx_Analysis_2017–06–05_v8_RNASeQCv1.1.9_gene_reads.gct.gz); and gender information was obtained from the file (GTEx_Analysis_v8_Annotations_SubjectPhenotypesDS.txt). The median (refmedian) and mean (refmean) of each gene were taken as read counts of the corresponding gene in the reference database. The medians of ratios analyzed using DESeq2 were used to normalize each sample (Anders and Huber, 2010). The analysis of differentially expressed genes was obtained by comparing data from the male (II-4) with his father (I-2), older sister (II-2), mother (I-1), and the database (GTEx database (v8)). The values of the read counts after normalization using DESeq2 are shown in Supplementary Material S1, and the codes for RNA-seq data analysis are provided in Supplementary Material S2. RNA-seq data were deposited on the Genome Sequence Archive for Human (https://ngdc.cncb.ac.cn/gsa-human/) with accession number HRA003790, and data on chromosomal microarray analysis were deposited on OMIX (https://ngdc.cncb.ac.cn/omix/) with accession number OMIX002784.
ResultsPatient characteristics and clinical observationIn a missed abortion of a woman (II-2) (Figure 1A), chromosomal microarray analysis (CMA) of aborted tissue (III-3, female fetus) was performed in other hospital, and duplication of Xq28 (arr [GRCh37] Xq28 (154109413–154228924) ×3) was found. As this region involved F8, the relatively high incidence of F8 inversion was further analyzed, and Inv22 was also found in the aborted tissue. Then, the woman (II-2) came to our hospital for genetic counseling on the reproductive risk of hemophilia A. As there was no hemophilia A patient in her family, we further analyzed the peripheral blood samples from the woman (II-2) by CMA and the F8-specific inversion panel. Also, we found duplication in the Xq28 region (arr [GRCh37] Xq28 (154120633–154228924) ×3) and Inv22. Because Inv22 is a common pathogenic variant of hemophilia A, the woman (II-2) preferred to perform genetic testing of her family to assess the pathogenicity of this complex variant. Blood coagulation factor VIII activities in all four family members (I-1, I-2, II-2, and II-4) were normal (Supplementary Material S3).
FIGURE 1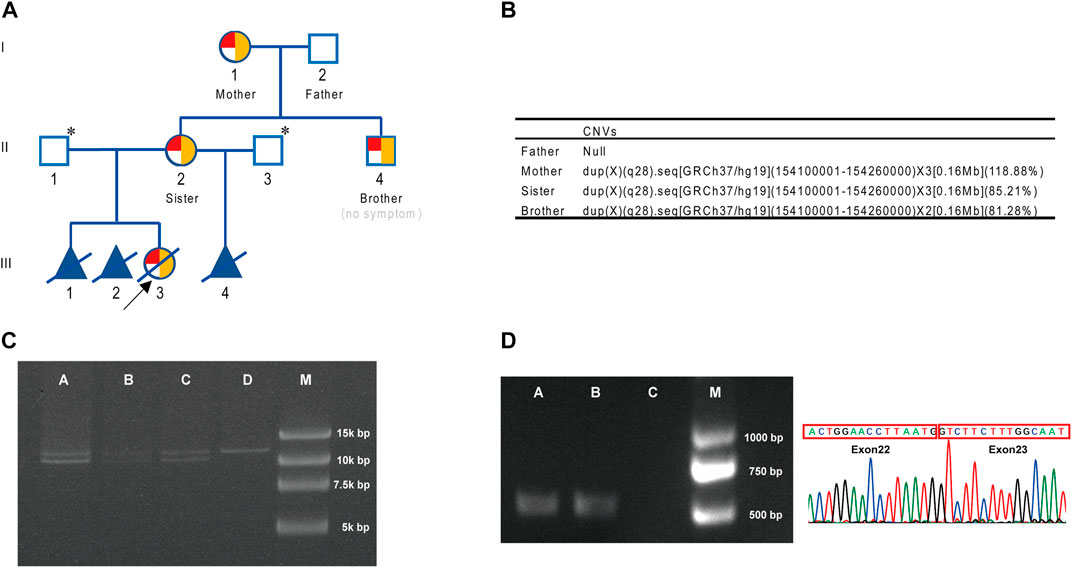
FIGURE 1. Pedigree and genetic analysis. (A) Genogram for the family (with CNV and inversion results). Red for inversion, 1/4 red circle for heterozygosis, 1/2 red circle for homozygosis, and yellow for duplication. * indicates that F8 gene analysis was not performed. (B) CNV results for the family. (C) Inv22 assay for the family. A, brother (II-4); B, sister (II-2); C, mother (I-1); D, father (I-2); M, marker. (D) Connection between exon 22 and exon 23 at the transcript level. A, brother (II-4); B, sister (II-2); C, blank; M, marker.
Genetic testingCNV analysis was conducted on the four members of this family, and it was found that his mother (I-1), sister (II-2), and the male subject (II-4) all carried a 0.16 Mb duplication of F8 gene ([GRCh37/hg19] chr.X: 154100001–154260000) (Figure 1B). The duplication region contained exon 1–exon 22 of F8 gene and partial of its flanking sequences (including partial intron 22 and 5′-UTR of F8). Inv22 analysis of the family members using the F8-specific inversion panel revealed that the mother (I-1) and the woman (II-2) both carried heterozygous Inv22, and her younger brother (II-4) was hemizygous, while her father (I-2) was normal (Figure 1C). Interestingly, the woman‘s younger brother (II-4) also carried this Inv22 of F8. At that time, he was a college student of 24 years old who usually liked playing basketball and never experienced joint swelling and pain, gingival bleeding, or unstoppable bleeding. That is to say he did not have a significant hemophilia A disease phenotype (blood coagulation factor VIII activities 69.1% (Supplementary Material S3)).
Gene expressionIn general, Inv22 of the F8 gene in males always results in an absent normal transcript, that is, there is no exon 22–exon 23-connected transcript (exon 23 is located after the breakpoint of the inversion). However, there were exon 22–exon 23-linked transcripts in this male (II-4) (Figure 1D), that is, there were normal F8 gene transcripts in this male. This result suggested that Inv22 might be located in the duplication region and did not affect the normal expression of F8.
RNA-seq analysis showed that the expression of F8 was very low in the blood of all the family members. But interestingly, the expression of C1QA in this male (II-4) as well as in his mother (I-1) and sister (II-2) was only half of that in his father (I-2) and normal control. This was also validated by Q-PCR (Figure 2).
FIGURE 2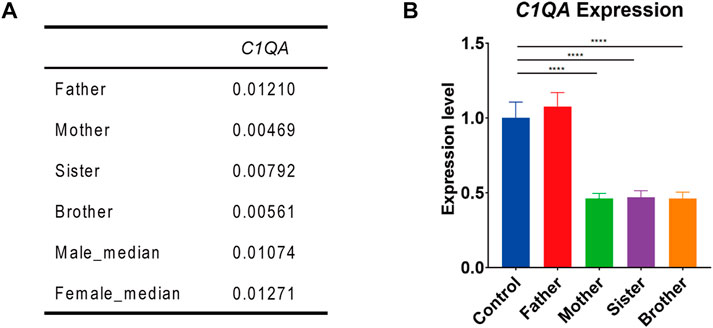
FIGURE 2. C1QA expression in the family members. (A) C1QA expression levels in four family members and reference databases (male_median and female_median) by RNA-seq analysis (values represented the relative expression to GAPDH). (B). Q-PCR confirmation of C1QA expression in four family members and normal control (one-way ANOVA and Dunnett’s multiple comparison test; ****, p < 0.0001).
DiscussionIn this case, we presented a male without obvious hemophilia A phenotype but hemizygous for complex Inv22 of the F8 gene, accompanied by a partial duplication of 0.16 Mb. This partial duplication and Inv22 in F8 were also found in his mother and sister. Considering that the exon 22–exon 23-linked transcripts and Inv22 both presented in the male (II-4), combined with the mechanism by which Inv22 occurs, we speculated that Inv22 occurred in the duplicated region, which was likely to be located upstream of F8 (Figure 3). By analyzing the RNA expression, we found that the expression of C1QA in his mother, sister, and the male subject was about only half of that in his father and normal population. This was the first report of such a complex structural variation of F8 in an asymptomatic male.
FIGURE 3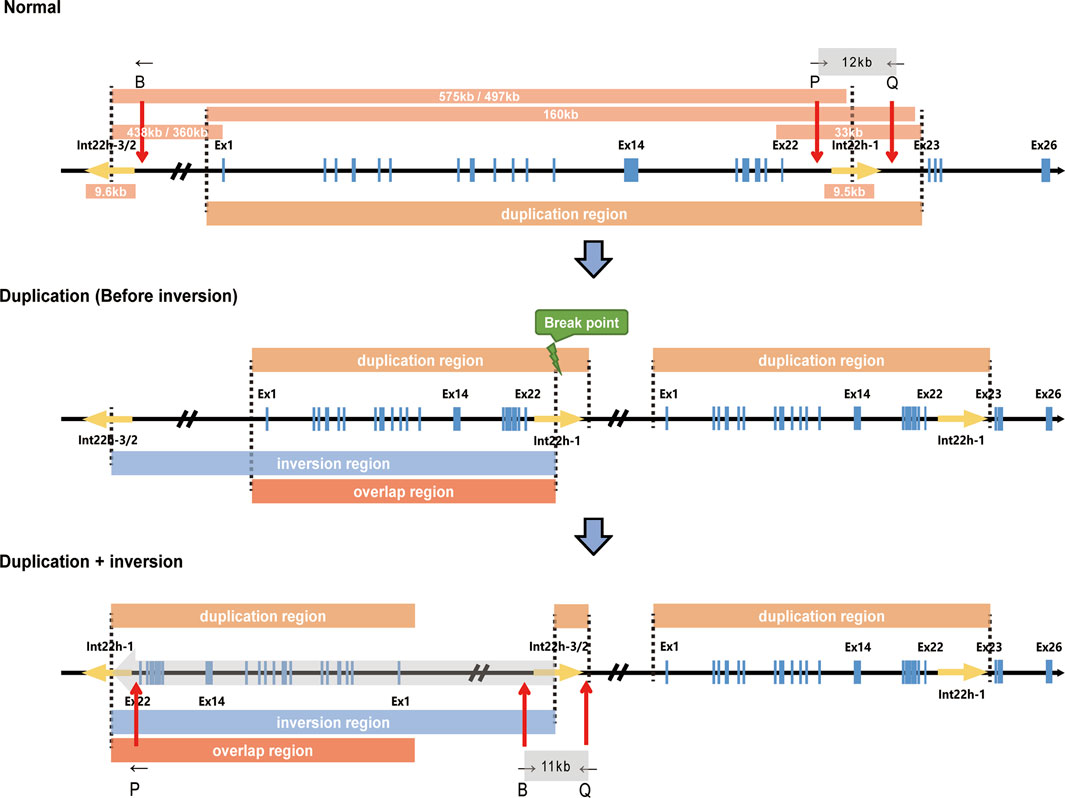
FIGURE 3. Schematic description of F8 duplication and inversion. The upper, middle, and lower panels represent the normal F8, the duplication of F8 (in which inversion has not occurred yet), and the duplication and further inversion of F8, respectively. The size of specific regions, duplication regions, inversion regions, and regions overlapping duplication and inversion are indicated. B, P, and Q show the relative positions of the primers used in Inv22 analysis. Ex1, Exon 1; Ex14, Exon 14; Ex22, Exon 22; Ex23, Exon 23; Ex26, Exon 26.
The most common variant of F8 is Inv22 (Antonarakis et al., 1995), and the remaining variants include Inv1, point mutations (including nonsense mutations), and small deletions (Bagnall et al., 2002; Oldenburg et al., 2004). In 2,401 patients with hemophilia type A (mild, moderate, and severe), the incidence of large duplications (>50bp) was about 0.46% (11/2,401), compared with Inv22 (24.1%), Inv1 (0.83%), missense mutation (46.4%), frameshift mutation (10.2%), nonsense mutation (6.2%), and splicing mutation (3.0%) (Johnsen et al., 2017). So far, only 52 HA patients with large duplications of F8 have been reported (Table 1). Specifically, six of these patients were combined with F8 inversion. Two patients (one male and one female) with Inv22, combined with exon 23–25 duplication, had no overlap between the inversion and the duplication region (Johnsen et al., 2017). One severe HA patient had overlapped duplication (intron 1–6) and inversion region (intron 1) in F8 (Sanna et al., 2013). Only Lannoy et al. (2015) reported similar inversion duplication within F8 intron 1 from three unrelated families with mild hemophilia A. In general, Inv1 of the F8 gene always caused severe hemophilia A, but in those probands, tandem inversion duplication (210 kb) did not completely disrupt the F8 expression and still retained a small number of normal mRNA transcripts. In contrast to mild hemophilia A, we reported that the FVIII factor activity reached 69.1% (normal range 70%–150%) in the asymptomatic male (II-4) with duplication and Inv22. Therefore, it was speculated that this structural variation not only brought no changes to the integrity of the open reading frame but also had no significant effect on the promoter of F8.
TABLE 1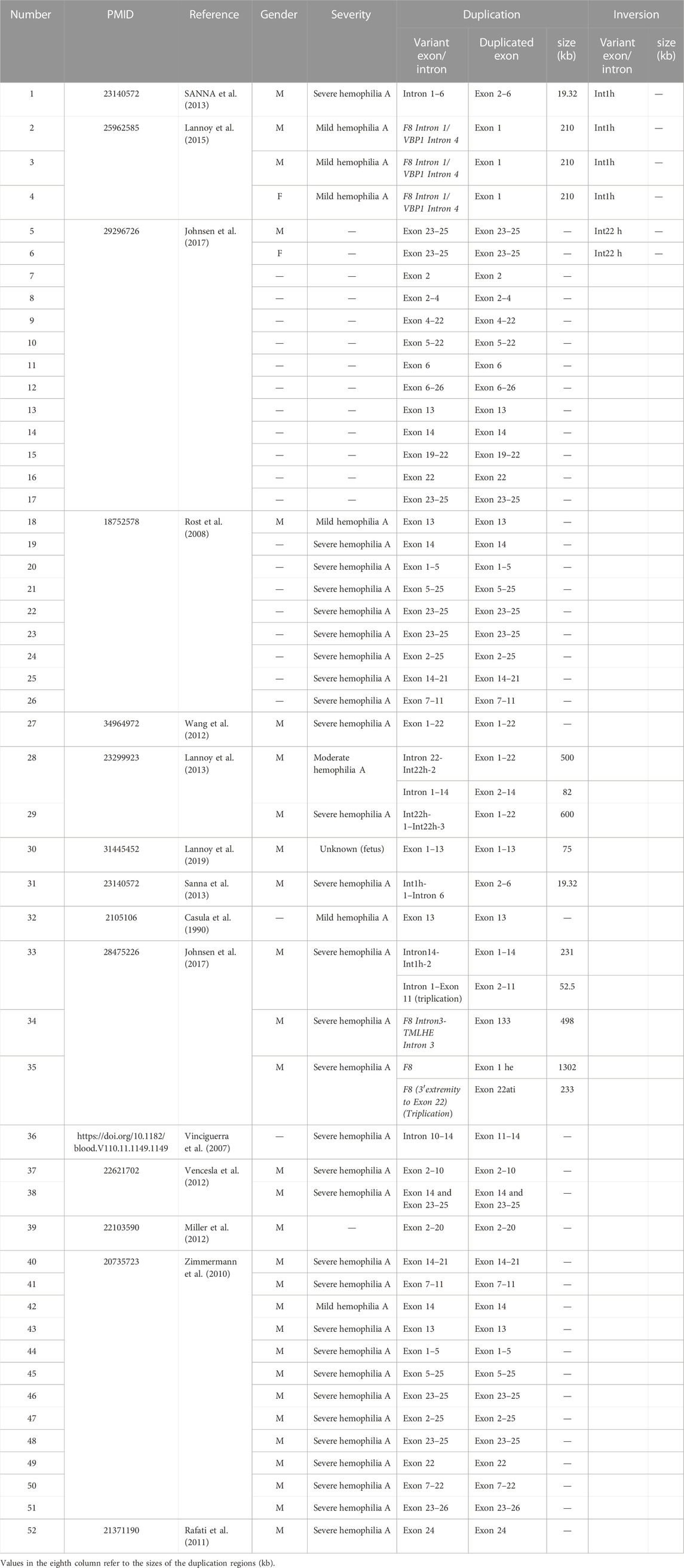
TABLE 1. Fifty-two HA patients with large duplications of F8.
C1qA is the A-chain peptide of C1q, which binds C1r and C1s to produce the first component of the serum complement system. The complement system is the humoral backbone of innate immune defense and is involved in many different processes, including antibacterial defense, clearance of immune complexes, and tissue regeneration. C1, the identifying unit of the complement system, is a complex with a size of 790 kDa, which contains three subcomponents: C1q, C1r, and C1s. C1q is the recognition component, and C1r and C1s are two proteases. The two copies of C1r and C1s combine to form a Ca2+-dependent tetramer, which together with C1q forms C1 (Gaboriaud et al., 2004). C1QA is a 3,216-bp gene and locates on chromosome 1, encoding 245 aa (NM_015991.4). It is widely expressed in the spleen, lymph node, whole blood, and 29 other tissues (https://www.genecards.org/cgi-bin/carddisp.pl?gene=C1QA&keywords=C1QA#expression). C1qA is associated with angiogenesis, innate immune response, osteosarcoma (OS), schizophrenia, hypertension, aging, and obesity. According to rat aortic ring assay, C1q induced permeability of the endothelial cell (EC) monolayer, stimulated proliferation and migration of the EC, and promoted tube formation and the sprouting of new vessels (Bossi et al., 2014). In innate immune response, C1qA interacted with various components of the RIG-I (retinoic acid-inducible gene-I) pathway and enhanced the RIG-I-VISA (virus-induced signaling adaptor)-mediated signaling pathway and TBK1 (TANK-binding kinase 1)-mediated activation of the interferon-β promoter (Wang et al., 2012). Rarely, people with a deficiency of the complement component C1q were prone to recurrent infections with polysaccharide-containing encapsulated microorganisms and a high prevalence of autoimmune diseases, mainly systemic lupus erythematosus (SLE) (Sun-Tan et al., 2010; Hayakawa et al., 2011). C1QA, C1QB, and C1QC expression levels were positively correlated with OS patient survival time and negatively correlated with the clinicopathological feature percent necrosis at definitive surgery (Chen et al., 2021). In schizophrenia, the gene expression of complement pathway activators (C1qA) and mediators (C3 or C4) was increased in the midbrain, especially in patients with inflammatory biotypes (Purves-Tyson et al., 2020). The loss of C1QA would also reduce hypertension-induced β-catenin signaling, proliferation of vascular smooth muscle cells, and pathological arterial remodeling (Sumida et al., 2015). During normal aging, the levels of the C1q protein in the mouse and human brain dramatically increased, and in certain tests of hippocampus-dependent behavior, C1q-deficient mice showed no cognitive or memory decline compared to their wild-type littermates (Stephan et al., 2013; Soto et al., 2015). In the diet-induced obesity mouse model, C1qA was necessary to cause damage to cerebral vasculature and white matter (Graham et al., 2020).
The expression of C1QA in this male (II-4) as well as in his mother (I-1) and sister (II-2) was only half of that in his father (I-2) and normal control. It is unknown whether this change in C1QA expression is directly related to the structural variation of F8, but we speculate that it may be at some risk in wound healing, recurrent infections, and even autoimmune diseases, but whether there will be related symptoms requires further follow-up.
The limitation to this study was the regulatory mechanisms of C1QA. The impact of F8 on C1QA expression and its association with this complex structural variant also required further investigation in animal models and cell models.
In conclusion, here, we presented an asymptomatic male with complex hemizygous Inv22 of the F8 gene, accompanied by a partial duplication of 0.16 Mb. This partial duplication and Inv22 in F8 were also found in his mother and sister. By RNA-seq analysis, we also found that the expression of C1QA in his mother, sister, and the male subject was about only half of that in his father and normal population. As far as we know, this is the first report of partial duplication and Inv22 in F8 in a male without hemophilia A phenotype. Therefore, caution is recommended in the use of conventional Inv22 or Inv1 assay for clinical prediction, especially with no family history of hemophilia A. Lastly, our study highlighted the complexity of the underlying molecular mechanisms in F8 structural variation in hemophilia A.
Data availability statementThe RNA-seq data presented in the study are deposited in the Genome Sequence Archive for human repository (https://ngdc.cncb.ac.cn/gsa-human/), accession number HRA003790. The data of chromosomal microarray analysis presented in the study are deposited in the OMIX repository (https://ngdc.cncb.ac.cn/omix/), accession number OMIX002784.
Ethics statementThe studies involving human participants were reviewed and approved by the Ethics Committee of The Third Affiliated Hospital of Guangzhou Medical University. The patients/participants provided their written informed consent to participate in this study.
Author contributionsSL, JH, and WH collected clinical data and blood samples and performed DNA extraction. LC, SR, XM, YW, and MZ conducted CNV analysis and RNA-seq. SL, JH, LC, SR, WH, XM, YW, and MZ collected the literature and wrote the manuscript. SL, JH, LC, LK, BL, and QL were responsible for the study design and guiding of the study implementation and revised the manuscript. All authors contributed to the article and approved the submitted version.
FundingThis project was funded by the Basic and Applied Basic Research Foundation of Guangdong (2021A1515220152).
AcknowledgmentsThe authors thank the family members for participating in this study.
Conflict of interestAuthors LC, SR, and LK were employed by Basecare Medical Device Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1098795/full#supplementary-material
Supplementary Material S1 | The values of the read counts (RNA-seq) after normalization using DESeq2.
Supplementary Material S2 | RNA-seq data analysis codes.
Supplementary Material S3 | Biochemical tests results of coagulation factor VIII.
ReferencesAnders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11 (10), R106. doi:10.1186/gb-2010-11-10-r106
PubMed Abstract | CrossRef Full Text | Google Scholar
Antonarakis, S. E., Rossiter, J. P., Young, M., Horst, J., de Moerloose, P., Sommer, S. S., et al. (1995). Factor VIII gene inversions in severe hemophilia A: Results of an international consortium study. Blood 86 (6), 2206–2212. doi:10.1182/blood.v86.6.2206.bloodjournal8662206
PubMed Abstract | CrossRef Full Text | Google Scholar
Bagnall, R. D., Waseem, N., Green, P. M., and Giannelli, F. (2002). Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 99 (1), 168–174. doi:10.1182/blood.v99.1.168
PubMed Abstract | CrossRef Full Text | Google Scholar
Bossi, F., Tripodo, C., Rizzi, L., Bulla, R., Agostinis, C., Guarnotta, C., et al. (2014). C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc. Natl. Acad. Sci. U. S. A. 111 (11), 4209–4214. doi:10.1073/pnas.1311968111
PubMed Abstract | CrossRef Full Text | Google Scholar
Casula, L., Murru, S., Pecorara, M., Ristaldi, M. S., Restagno, G., Mancuso, G., et al. (1990). Recurrent mutations and three novelrearrangements in the factor VIII gene of hemophilia A patients of Italiandescent. Blood 75 (3), 662–670.
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, L. H., Liu, J. F., Lu, Y., He, X. Y., Zhang, C., and Zhou, H. H. (2021). Complement C1q (C1qA, C1qB, and C1qC) may Be a potential prognostic factor and an index of tumor microenvironment remodeling in osteosarcoma. Front. Oncol. 11, 642144. doi:10.3389/fonc.2021.642144
PubMed Abstract | CrossRef Full Text | Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Star: Ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
PubMed Abstract | CrossRef Full Text | Google Scholar
Freije, D., and Schlessinger, D. (1992). A 1.6-Mb contig of yeast artificial chromosomes around the human factor VIII gene reveals three regions homologous to probes for the DXS115 locus and two for the DXYS64 locus. Am. J. Hum. Genet. 51 (1), 66–80.
PubMed Abstract | Google Scholar
Gaboriaud, C., Thielens, N. M., Gregory, L. A., Rossi, V., Fontecilla-Camps, J. C., and Arlaud, G. J. (2004). Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol. 25 (7), 368–373. doi:10.1016/j.it.2004.04.008
PubMed Abstract | CrossRef Full Text | Google Scholar
Gitschier, J., Wood, W. I., Goralka, T. M., Wion, K. L., Chen, E. Y., Eaton, D. H., et al. (1984). Characterization of the human factor VIII gene. Nature 312 (5992), 326–330. doi:10.1038/312326a0
PubMed Abstract | CrossRef Full Text | Google Scholar
Gouw, S. C., van den Berg, H. M., Oldenburg, J., Astermark, J., de Groot, P. G., Margaglione, M., et al. (2012). F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 119 (12), 2922–2934. doi:10.1182/blood-2011-09-379453
PubMed Abstract | CrossRef Full Text | Google Scholar
Graham, L. C., Kocalis, H. E., Soto, I., and Howell, G. R. (2020). Deficiency of complement component C1Q prevents cerebrovascular damage and white matter loss in a mouse model of chronic obesity. eNeuro 7 (3), ENEURO.0057–20.2020. doi:10.1523/ENEURO.0057-20.2020
PubMed Abstract | CrossRef Full Text | Google Scholar
Hayakawa, J., Migita, M., Ueda, T., Itoh, Y., and Fukunaga, Y. (2011). Infantile case of early manifestation of SLE-like symptoms in complete C1q deficiency. J. Nippon. Med. Sch. 78 (5), 322–328. doi:10.1272/jnms.78.322
PubMed Abstract | CrossRef Full Text | Google Scholar
Johnsen, J. M., Fletcher, S. N., Huston, H., Roberge, S., Martin, B. K., Kircher, M., et al. (2017). Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the My Life, Our Future initiative. Blood Adv. 1 (13), 824–834. doi:10.1182/bloodadvances.2016002923
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakich, D., Kazazian, H. H., Antonarakis, S. E., and Gitschier, J. (1993). Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat. Genet. 5 (3), 236–241. doi:10.1038/ng1193-236
PubMed Abstract | CrossRef Full Text | Google Scholar
Lannoy, N., Bandelier, C., Grisart, B., Reginster, M., Ronge-Collard, E., Vikkula, M., et al. (2015). Tandem inversion duplication within F8 Intron 1 associated with mild haemophilia A. Haemophilia 21 (4), 516–522. doi:10.1111/hae.12675
PubMed Abstract | CrossRef Full Text | Google Scholar
Lannoy, N., Grisart, B., Eeckhoudt, S., Verellen-Dumoulin, C., Lambert, C., Vikkula, M., et al. (2013). Intron 22 homologous regions are implicated in exons 1-22 duplications of the F8 gene. Eur. J. Hum. Genet. 21 (9), 970, doi:10.1038/ejhg.2012.275
PubMed Abstract | CrossRef Full Text | Google Scholar
Lannoy, N., and Hermans, C. (2016). Principles of genetic variations and molecular diseases: Applications in hemophilia A. Crit. Rev. Oncol. Hematol 104, 1–8. doi:10.1016/j.critrevonc.2016.04.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Lannoy, N., Lambert, C., Van Damme, A., and Hermans, C. (2019). Incidental finding of unreported large duplication in F8 gene during prenatal analysis: Which management for genetic counselling?, Thromb. Res 182, 39, doi:10.1016/j.thromres.2019.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Lenting, P. J., van Mourik, J. A., and Mertens, K. (1998). The life cycle of coagulation factor VIII in view of its structure and function. Blood 92 (11), 3983–3996. doi:10.1182/blood.v92.11.3983
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, W., Lu, J., Zhang, J., Li, R., Lin, S., Zhang, Y., et al. (2020). A consensus recommendation for the interpretation and reporting of copy number variation and regions of homozygosity in prenatal genetic diagnosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 37 (7), 701–708. doi:10.3760/cma.j.issn.1003-9406.2020.07.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Lu, D., Xue, Y., Song, B., Liu, N., Xie, Y., Cheng, Y., et al. (2020). Generation of induced pluripotent stem cell GZLSL-i001-A derived from urine-derived cells of Hemophilia A patient with Inv22 mutation. Stem Cell Res. 49, 102053. doi:10.1016/j.scr.2020.102053
PubMed Abstract | CrossRef Full Text | Google Scholar
Mahmoud Abu Arra, C., Samarah, F., and Sudqi Abu Hasan, N. (2020). Factor VIII intron 22 inversion in severe hemophilia A patients in Palestine. Scientifica Cairo2020, 3428648. doi:10.1155/2020/3428648
PubMed Abstract | CrossRef Full Text | Google Scholar
Miller, C. H., Benson, J., Ellingsen, D., Driggers, J., Payne, A., Kelly, F. M., et al. (2012). F8 and F9 mutations in US haemophiliapatients: correlation with history of inhibitor and race/ethnicity. Haemophilia 18 (3), 375–382. doi:10.1111/j.1365-2516.2011.02700.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Oldenburg, J., Ananyeva, N. M., and Saenko, E. L. (2004). Molecular basis of haemophilia A. Haemophilia 10 (4), 133–139. doi:10.1111/j.1365-2516.2004.01005.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Purves-Tyson, T. D., Robinson, K., Brown, A. M., Boerrigter, D., Cai, H. Q., Weissleder, C., et al. (2020). Increased macrophages and C1qA, C3, C4 transcripts in the midbrain of people with schizophrenia. Front. Immunol. 11, 2002. doi:10.3389/fimmu.2020.02002
PubMed Abstract | CrossRef Full Text | Google Scholar
Rafati, M., Ravanbod, S., Hoseini, A., Rassoulzadegan, M., Jazebi, M., Enayat, M. S., et al. (2011). Identification of ten largedeletions and one duplication in the F8 gene of eleven unrelated Iranian severehaemophilia A families using the multiplex ligation-dependent probeamplification technique. Haemophilia 17 (4), 705–707. doi:10.1111/j.1365-2516.2010.02476.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Rost, S., Loffler, S., Pavlova, A., Muller, C.R., and Oldenburg, J. (2008). Detection of large duplications within the factor VIII gene by MLPA. J Thromb Haemost 6 (11), 1996–1999. doi:10.1111/j.1538-7836.2008.03125.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Sanna, V., Ceglia, C., Tarsitano, M., Lombardo, B., Coppola, A., Zarrilli, F., et al. (2013). Aberrant F8 gene intron 1 inversion with concomitant duplication and deletion in a severe hemophilia A patient from Southern Italy. J. Thromb. Haemost. 11 (1), 195–197. doi:10.1111/jth.12061
PubMed Abstract | CrossRef Full Text | Google Scholar
Soto, I., Graham, L. C., Richter, H. J., Simeone, S. N., Radell, J. E., Grabowska, W., et al. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 13 (10), e1002279. doi:10.1371/journal.pbio.1002279
PubMed Abstract | CrossRef Full Text | Google Scholar
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33 (33), 13460–13474. doi:10.1523/JNEUROSCI.1333-13.2013
PubMed Abstract | CrossRef Full Text | Google Scholar
Sumida, T., Naito, A. T., Nomura, S., Nakagawa, A., Higo, T., Hashimoto, A., et al. (2015). Complement C1q-induced activation of beta-catenin signalling causes hypertensive arterial remodelling. Nat. Commun. 6, 6241. doi:10.1038/ncomms7241
PubMed Abstract | CrossRef Full Text | Google Scholar
Sun-Tan, C., Ozgur, T. T., Kilinc, G., Topaloglu, R., Gokoz, O., Ersoy-Evans, S., et al. (2010). Hereditary C1q deficiency: A new family with C1qA deficiency. Turk J. Pediatr. 52 (2), 184–186.
PubMed Abstract | Google Scholar
Vinciguerra, C., Goudemand, J., Zawadzki, C., Pellecchia, D., and Negrier, C. (2007). Am. J. Hematol. J. Instrum. doi:10.1182/blood.V110.11.1149.1149
CrossRef Full Text | Google Scholar
Vencesla, A., Baena, M., Garrido, R. P., Nunez, R., Velasco, F., Rosell, J., et al. (2012). F8 gene dosage defects in atypical patients withsevere haemophilia A. Haemophilia 18 (5), 708–713. doi:10.1111/j.1365-2516.2012.02818.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, Y., Tong, X., Zhang, J., and Ye, X. (2012). The complement C1qA enhances retinoic acid-inducible gene-I-mediated immune signalling. Immunology 136 (1), 78–85. doi:10.1111/j.1365-2567.2012.03561.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Zimmermann, M. A., Oldenburg, J., Muller, C. R., and Rost, S. (2010). Characterization of duplication breakpoints in the factor VIII gene. J. Thromb. Haemost. 8 (12), 2696–2704. doi:10.1111/j.1538-7836.2010.04040.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Keywords: F8, hemophilia A, intron 22 inversion, F8 duplication, C1qA
Citation: Li S, He J, Chu L, Ren S, He W, Ma X, Wang Y, Zhang M, Kong L, Liang B and Li Q (2023) F8 gene inversion and duplication cause no obvious hemophilia A phenotype. Front. Genet. 14:1098795. doi: 10.3389/fgene.2023.1098795
Received: 15 November 2022; Accepted: 23 January 2023;Published: 09 February 2023.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Enrique Medina-Acosta, State University of the North Fluminense Darcy Ribeiro, BrazilMing Qi, Zhejiang University, ChinaCopyright © 2023 Li, He, Chu, Ren, He, Ma, Wang, Zhang, Kong, Liang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Li, 81292522@163.com; Bo Liang, boliang880@sjtu.edu.cn
†These authors have contributed equally to this work